Abstract
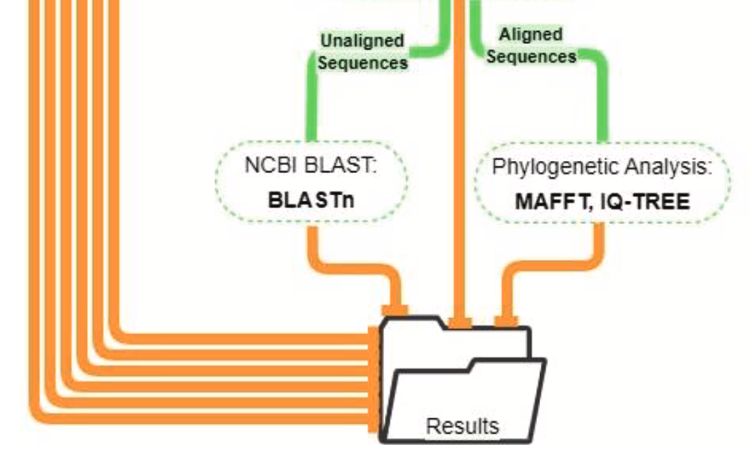
The Centers for Disease Control and Prevention's national tick and tick-borne pathogen surveillance program collects information to better understand the regional distribution, prevalence, and exposure risk of host-seeking medically important ticks in the United States. A recently developed next generation sequencing (NGS) targeted multiplex PCR amplicon sequencing (MPAS) assay has enhanced the detection capabilities for Ixodes-associated human pathogens found in Ixodes scapularis and Ixodes pacificus ticks compared to the routinely used real-time PCR assay. To operationalize the MPAS assay for the large number of tick surveillance submissions processed each year, a reproducible high throughput bioinformatics pipeline is needed. We describe the development and validation of the MPAS pipeline, a bioinformatics pipeline that identifies and summarizes amplicon sequences produced by the MPAS assay. This pipeline is portable and reproducible across different computing environments, and flexible by allowing modifications to input parameters, assay primer and reference sequences. The automation of the summary report, BLAST report, and phylogenetic analysis reduces the amount of time needed for downstream analysis. To validate this pipeline, we compared the analysis of a MPAS assay dataset consisting of 175 I. scapularis nymphs with the MPAS pipeline and previously published results analyzed with a CLC Genomic Workbench workflow. The MPAS pipeline identified the same number of positive ticks for Anaplasma phagocytophilum and Babesia species as the original analysis, but the MPAS pipeline provided enhanced sequencing resolution of Borrelia burgdorferi sensu lato co-infected samples. The reproducibility, flexibility, analysis automation, and improved sequence resolution of the MPAS pipeline make it well suited for a high throughput tick pathogen surveillance program.