A paper published online on Jan 20, 2020 in the Journal of Medical Virology, titled Homologous recombination within the spike glycoprotein of the newly identified coronavirus 2019-nCoV may boost cross-species transmission from snake to human raises the intriguing possibility that a new coronavirus of substantial public health concern, 2019-nCoV, may have originated in snakes.
The authors make two primary claims. First, that 2019-nCoV is a recombinant virus. Second, that "...our evolutionary analysis suggest for the first time that snake is the most probable wildlife animal reservoir for the 2019-nCoV."
Recombination occurs when two viruses exchange genetic material. I'm not going to evaluate the claim that this virus may be recombinant.
I will however, consider the evidence that this virus may have originally been transmitted to humans from snakes. As I describe below, there is no direct evidence to support this claim, and the indirect evidence is flimsy.
Many pathogens can be transmitted from other animals to humans. Snakes were apparently one of the types of animals sold at the seafood market that has been linked to many of the initial human cases of 2019-nCoV. So it is plausible that this virus could have been from snakes.
There is no direct evidence that this type of coronavirus infects snakes. The authors do not identify a similar virus in snakes, and no similar coronaviruses have been identified previously in snakes.
(My lab has studied respiratory viruses of snakes that are belong to the same umbrella virus group as 2019-nCoV, but these are distant relatives).
The main line of evidence the authors present to support the claim that this virus may have originated in snakes is indirect, and based on codon usage patterns. Codons are DNA base triplets that encode a single amino acid in a protein. The same amino acid may be encoded by different codons, so there is flexibility in how a particular protein sequence could be encoded. Different organisms tend to use codons in a characteristic way. Viruses that matched their codon usage to that of their host could have an evolutionary advantage.
So if, as seems likely, 2019-nCoV was recently transmitted to humans from other animals, then it might be possible to discern the original host by comparing codon usage patterns of the virus to candidate hosts.
The authors compare the relative codon usage of 2019-nCoV with several candidate hosts, corresponding to some of the animals that were sold in the seafood market that has been linked to this outbreak and to the most closely related cornavirus to 2019-nCoV, bat-SL-CoVZC45, which was identified in a bat in 2017. The authors then calcualte the squared Euclidian distance between as a measure of how similar these codon usage patterns are between the various species. The results are shown in figure 3 of the paper:
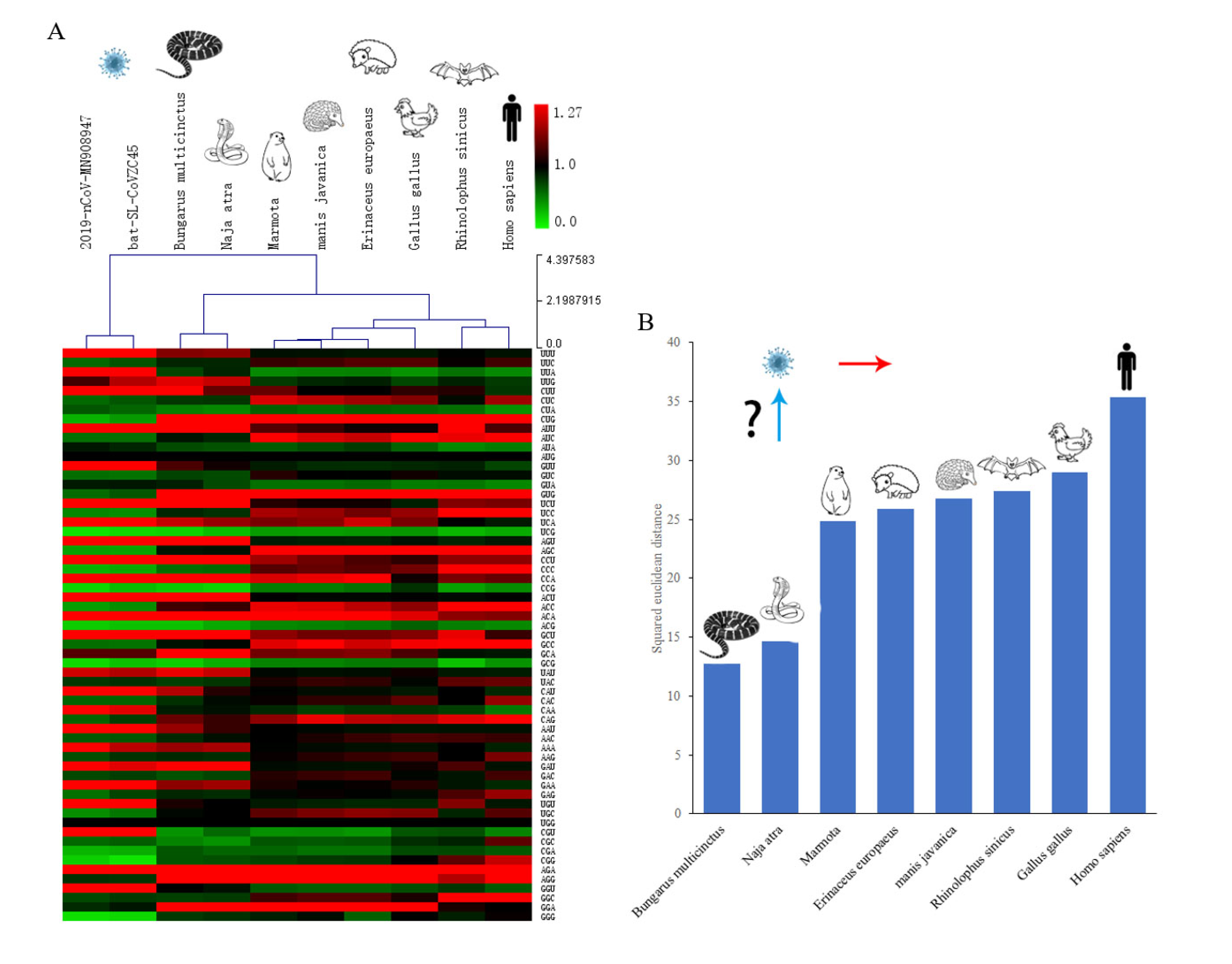
The "heatmap" in panel A depicts the frequencies with which codons are used in different organisms. Each column of the table is one organism. Each row is one of 61 possible codons. A redder color indicates that a particular codon is used more often in that organism. A green color means that codon is used less frequently. The heirarchical branching at the top of the figure indicates the extent to which these patterns are similar in different species. You can see that the two similar viruses (the two left-most columns) have closely related codon usage patterns, as expected. The two snakes have relatively similar patterns, and the mammals have similar patterns.
The claim that snakes are the "most probable wildlife" origin of 2019-nCoV rest on the bar graph shown in panel B which shows the Euclidian distance between the codon usage frequency vectors of 2019-nCoV and the various possible hosts. The distance between 2019-nCoV and the 2 snakes is ~12-15, whereas the distance between that virus and the mammals is >25. At first glance, this seems convincing.
But consider the case of the other coronavirus analyzed, bat-SL-CoVZC45. This virus was isolated from a Rhinolophus sinicus bat in China in 2017. It's codon usage pattern is essentially identical to that of 2019-nCoV. Consequently, the distances between it's codon usage pattern and the candidate hosts will be essentially identical to those shown in panel B. By the logic presented here, the bat-SL-CoVZC45 virus, from a Rhinolophus sinicus bat, is more likely to have originated from a snake than from a Rhinolophus sinicus bat (which is one of the mammalian species analyzed).
It is possible to imagine a scenario where previously unknown snake coronaviruses independently transmitted to bats and to humans. But in my opinion, a simpler and more likely explanation is that this kind of codon usage analysis has limited predictive value to pinpoint a virus's exact host in a situation like this.
It remains possible that snakes are the source of 2019-nCoV, but the data presented here is not convincing evidence of that.